

The Daily Free Press recounts the HUBWeek event in which Center Director Bruce Rosen and medical illustrator Danny Quirk spoke about the intersectionality of human anatomy and visual art.
Computational Method Opens New Avenues For Heritability Analysis
February 10, 2015
 |
|
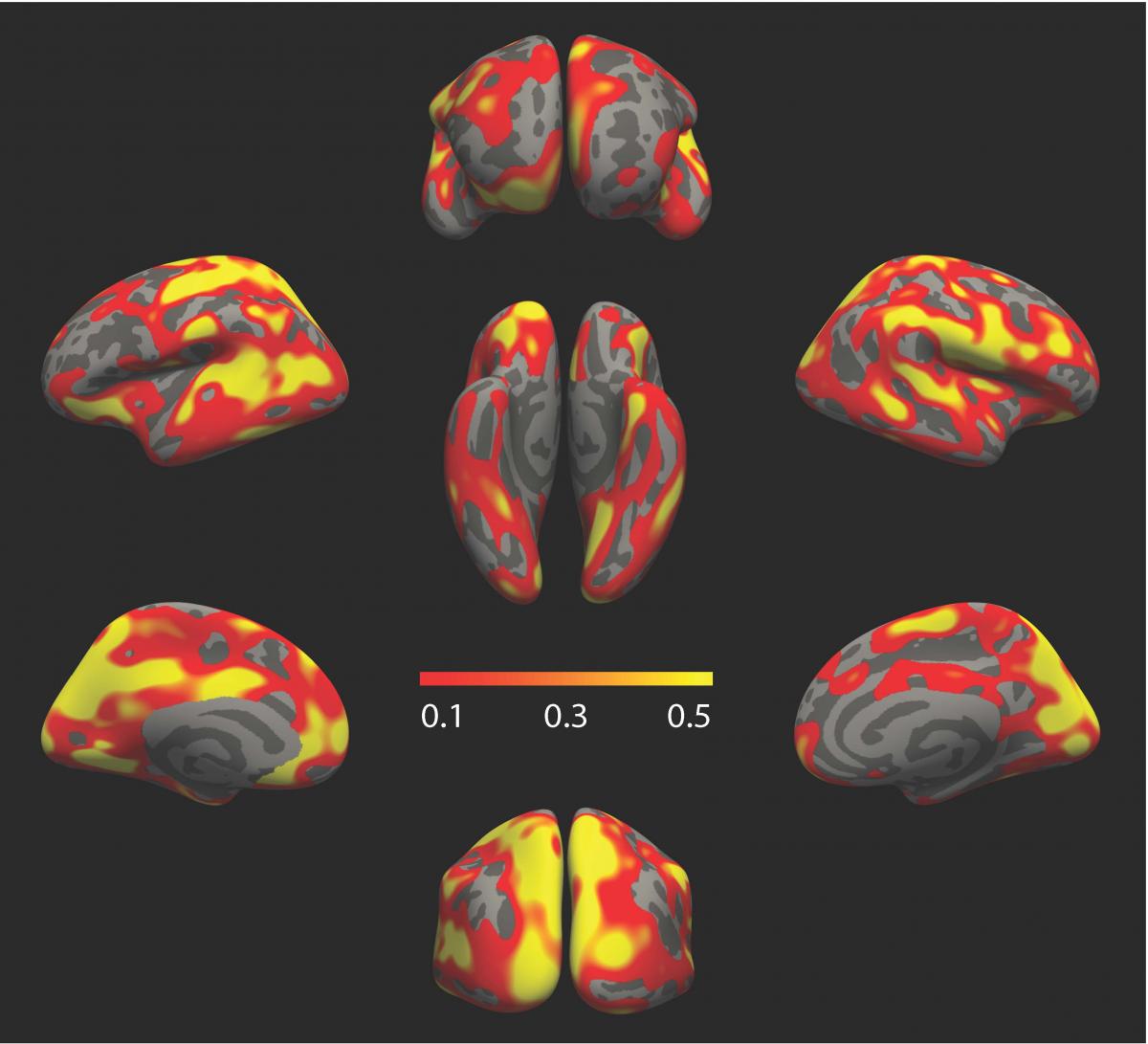
Martinos Center researchers have reported a new computational tool - called MEGHA - that enables analysis of the heritability of millions of clinical phenotypes. Shown here is a surface map of heritability of cortical thickness measures constructed using the tool. (Image courtesy of Tian Ge and Mert Sabuncu.) |
The Martinos Center’s Mert Sabuncu and Tian Ge and colleagues have reported a novel computational tool with which to measure the heritability – or genetic contribution – of millions of clinical phenotypes. They described the tool, Massively Expedited Genome-wide Heritability Analysis (MEGHA), in a Proceedings of the National Academy of Sciences (PNAS) paper published this week.
Recent years have seen the rise of the era of data-driven medicine. Researchers and clinicians now collect tremendous amounts of data on patients, including brain scans with up to millions of voxel-level measurements, electronic health record data, lab and clinical test results, and more. These data provide rich descriptions of the underlying biology and can tell us a great deal about disease pathology.
This is significant for those studying disease. “Many clinical conditions that we are interested in studying – with the hope that we can prevent, manage, or cure these conditions – are known to be caused by a combination of genetic and environmental factors,” said Sabuncu, an Assistant Professor in the Martinos Center and a research affiliate at MIT's Computer Science and Artificial Intelligence Lab (CSAIL). “Understanding the genetic underpinnings can therefore be important to reveal the biological mechanisms that lead to disease, design therapeutic targets, and/or deploy personalized medicine.”
Heritability analysis contributes to such understandings by quantifying the extent to which genetic factors influence the phenotypic variation we observe across individuals. However, the twin or pedigree data classically used to estimate heritability can be impractical and prohibitively expensive to collect. MEGHA addresses this limitation by enabling analysis of genotype data from unrelated individuals – which is far easier to obtain – to quantify the heritability of potentially millions of clinical phenotypes. In this way, it could help to advance a wide range of genetics-based studies.
“This tool, we believe, will enable researchers to prioritize their resources for studying the genetic underpinnings of phenotypes by focusing on the more heritable (and thus more promising) variables,” Sabuncu said.
Having reported MEGHA, the researchers are now planning to apply it to novel large-scale datasets. “We are going to construct high-resolution heritability profiles for a variety of image-wide measurements in healthy subjects and in the context of different neuropsychiatric disorders,” said Tian Ge, a postdoctoral fellow in Sabuncu’s lab and the first author of and primary contributor to the PNAS study.
At the same time, they are looking into extending the tool to estimate the proportion of shared genetic causes between pairs of phenotypes. This will allow researchers to conduct genetics-based studies of the relationship between clinical phenotypes.
Other authors of the study include Thomas Nichols, Phil Lee, Avram Holmes, the Martinos Center's Joshua Roffman, the Martinos Center's Randy Buckner, and Jordan Smoller.